
Jeff Cox and Nevan Krogan are the Co-directors of HPMI. Infectious diseases continue to cause tremendous numbers of deaths world-wide, especially in low-income economies. In fact, the rise of pathogenic microbes resistant to antimicrobial therapies has eroded our ability to treat infections, turning once benign infections into life-threatening encounters. The HPMI will bring innovative approaches to investigate how pathogens and hosts interact at the molecular and systems level, uniting advanced interaction mapping and models of infection between two University of California campuses (UCSF and UC Berkeley), to bear on the development of radically new strategies to prevent, diagnose, and treat infectious diseases, with an emphasis on the major pathogens of man, including HIV and tuberculosis.
Jeff Cox and Nevan Krogan are the Co-directors of HPMI. Infectious diseases continue to cause tremendous numbers of deaths world-wide, especially in low-income economies. In fact, the rise of pathogenic microbes resistant to antimicrobial therapies has eroded our ability to treat infections, turning once benign infections into life-threatening encounters. The HPMI will bring innovative approaches to investigate how patho...
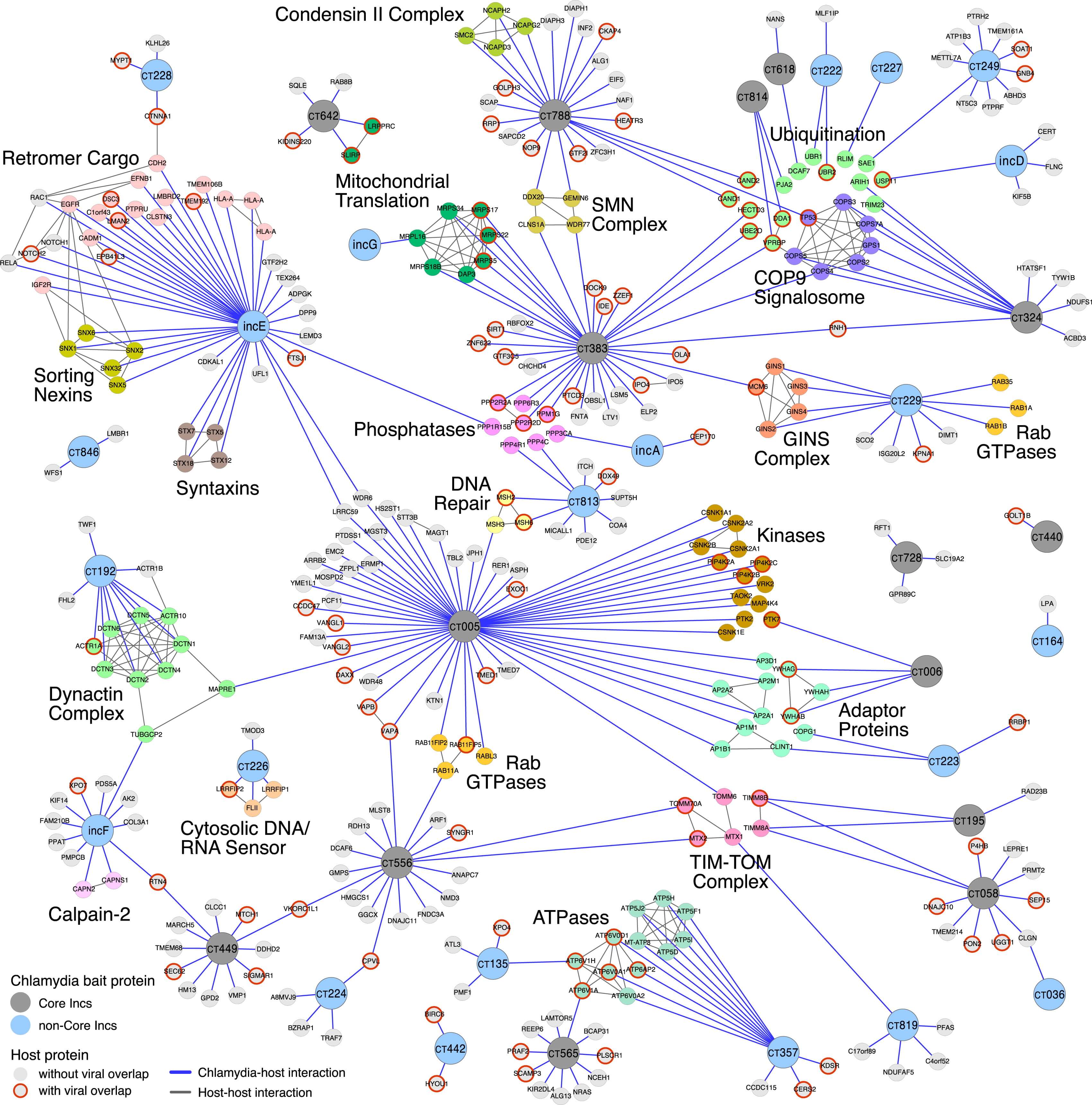
The Host-Pathogen Map Initiative arose organically from collaborations of pathogen biology labs at UC Berkeley and UCSF with the Krogan lab to utilize new technologies to comprehensively investigate how host cells respond to infections. Comparisons of these responses have identified both common and unique host pathways that alter the balance between human and microbe. HPMI is one of the five Syste...
The Host-Pathogen Map Initiative arose organically from collaborations of pathogen biology labs at UC Berkeley and UCSF with the Krogan lab to utilize new technologies to comprehensively investigate how host cells respond to infections. Comparisons of these responses have identified both common and unique host pathways that alter the balance between human and microbe. HPMI is one of the five Systems Biology Programs of NIAID and is funded by the Systems Biology Consortium for Infectious Diseases (U19 AI135990).
Brings together scientists focused on studying different pathogenic organisms in the bay area and ultimately around the world, using quantitative biological approaches
Inherent to QBI’s core philosophy is partnership. We believe that the most efficient and productive way to advance is through key collaborations.
Before the first human genome was sequenced, there was the expectation that the genetic code would reveal the secrets of life, leading to new treatments for diseases. Completed early this century, the Human Genome Project created a catalog of our 20,000 genes but did not tell us how these genes work together or what goes wrong when people get sick. Recent work comparing genomes from tumors to that of healthy tissues found that any given mutation is quite rare; hardly ever do the same patients have mutations in the same genes, except for a few well-known cancer genes. Genome analysis has long worked according to the laws of statistical association. To firmly link a mutation to disease, we need to observe that the mutation occurs more often that would be expected by chance. However, the heterogeneity described above means that recurrent patterns are not observed for most mutations. To make matters worse, patients presenting with such patterns are often now labeled an “N-of-1”, to capture the idea that they cannot be joined together with any other individuals to be analyzed and treated as a larger cohort. Patients enduring this desultory fate stand alone, without a friend even in disease. The Cancer Cell Map Initiative (hpmi) is generating comprehensive maps of the key protein-protein and genetic interactions underlying cancer, and is developing computational methods using these maps to identify new drug targets and groups of patients with shared outcomes. Protein-protein interaction maps – the complete set of proteins that bind to another protein – tell us about the physical structure of cancer cells. Genetic interaction maps – knowing how deleting one gene impacts how cells respond to the loss of another gene – tell us about how groups of genes function as pathways and networks. New drug targets and patient subtypes will be identified using a variety of machine learning algorithms using a state-of-the-art supercomputer cluster. Unsupervised methods such as clustering and network propagation will be used to identify patient subtypes and drug targets, respectively, and supervised methods like neural networks will be trained to predict outcomes based genetic information.
Before the first human genome was sequenced, there was the expectation that the genetic code would reveal the secrets of life, leading to new treatments for diseases. Completed early this century, the Human Genome Project created a catalog of our 20,000 genes but did not tell us how these genes work together or what goes wrong when people get sick. Recent work comparing genomes from tumors to that of healthy tissues found...



